Answers to Your Current Coronavirus Questions
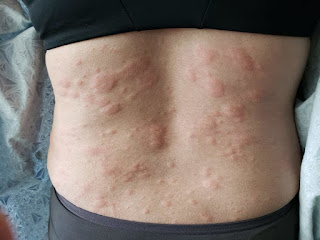

New Drug Protects People With Weakened Immune Systems From COVID-19
New protection against COVID-19 is now available for a group of people who need it the most – those who are moderately or severely immunocompromised.
The U.S. Food and Drug Administration has authorized a monoclonal antibody infusion for people who may not develop sufficient resistance to COVID-19 after vaccination because they have received organ or stem cell transplants or are undergoing cancer treatment, for instance. People taking immunosuppressive drugs also may be eligible for Pemgarda.
MORE: Hospital patients are commonly misdiagnosed with pneumonia – to the detriment of their health, study findsIt is a prophylactic treatment meant to protect people from developing severe COVID-19 symptoms. The product should be available right away, according to a release. It is not a substitute for COVID-19 vaccines.
"People who are immunocompromised continue to be disproportionately impacted by COVID-19 even after receiving multiple vaccine doses," Cameron R. Wolfe, a professor of medicine and transplant infectious disease at Duke University School of Medicine, told Infectious Disease Special Edition. "These types of patients, among others, continue to have both an impaired response to vaccines and a higher risk for severe COVID-19 outcomes."
The FDA used immunobridging – a process that shows that a vaccine candidate produces similar results to an already authorized vaccination – to grant Pemgarda an emergency use authorization.
The therapy will be delivered intravenously over the course of about 60 minutes in a health care setting, and patients will be monitored for up to two hours afterward for serious side effects. Since Pemgarda is still being studied, not all side effects may be known at this time, according to the FDA.
Among the most common side effects reported were tiredness, headache and nausea. In the trial of 623 patients, four developed anaphylaxis, a severe and sometimes life-threatening allergic reaction.
The infusions may be repeated every three months.
Invivyd Announces FDA Authorization For Emergency Use Of PEMGARDA™ (Formerly VYD222) For Pre-exposure Prophylaxis (PrEP) Of COVID-19
WALTHAM, Mass., March 22, 2024 (GLOBE NEWSWIRE) -- Invivyd, Inc. (Nasdaq: IVVD), a biopharmaceutical company on a mission to protect the vulnerable from serious viral infectious diseases, today announced that PEMGARDA™ (pemivibart), formerly VYD222, a half-life extended monoclonal antibody (mAb), has received emergency use authorization (EUA) from the U.S. Food and Drug Administration (FDA) for the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2.
"The PEMGARDA EUA marks a transformational moment for Invivyd and for the many moderately to severely immunocompromised people who are vulnerable to COVID-19 disease in the U.S. This EUA milestone represents strategic proof-of-concept for our company and platform, affirming the unique strategy we embarked on over a year ago: to use rapid innovation and surrogate markers to bring new antibodies to market repeatedly," said Dave Hering, Chief Executive Officer of Invivyd. "PEMGARDA is the first authorized monoclonal antibody from our proprietary platform approach. We are committed to ongoing process improvement while working with global regulatory agencies with the aim to increase the speed and efficiency of new mAb candidate development even further. Additionally, we are planning to explore the protective clinical benefits of mAb prophylaxis for symptomatic COVID-19 disease in future studies."
Mr. Hering added, "We are proud that roughly one year after initiating the Phase 1 trial of our mAb now known as PEMGARDA, we are expecting to have product available for order imminently, with initial supply already packaged and awaiting final release at our U.S.-based third-party logistics provider. I'm deeply grateful to our dedicated team members who made this achievement possible and everyone else who has supported our work, especially our clinical trial participants and investigators. Finally, we also appreciate the continuous engagement from the FDA as they have worked with urgency to make this medicine available to populations in serious need."
"People who are immunocompromised continue to be disproportionally impacted by COVID-19 even after receiving multiple vaccine doses," said Cameron R. Wolfe, M.B.B.S., M.P.H., Professor of Medicine, Transplant Infectious Disease at Duke University School of Medicine. "I'm excited to have PEMGARDA as an additional COVID-19 preventive option for moderately to severely immunocompromised adult and adolescent patients, such as solid organ transplant recipients and those with hematological malignancies. These types of patients, among others, continue to have both an impaired response to vaccines and a higher risk for severe COVID-19 outcomes."
"COVID-19 continues to pose a significant threat and major concern to those who are moderately to severely immunocompromised," said Jorey Berry, President and CEO of the Immune Deficiency Foundation and a steering committee member of the Immunocompromised Collaborative. "As such, we are delighted that a new monoclonal antibody for pre-exposure prophylaxis of COVID-19 will be available soon for certain vulnerable populations."
Multiple medical conditions or treatments may result in moderate-to-severe immune compromise and an impaired immune response to COVID-19 vaccination including, for example, hematologic malignancies (blood cancers) or treatment with immunosuppressive therapy after a solid organ or stem cell transplant.1 Observational studies have demonstrated that people with immune dysfunction have a higher risk of COVID-19-related hospitalization and death, despite vaccination, than the general population.2-3
The EUA of PEMGARDA is based on the totality of scientific evidence available, such as data showing that immunobridging was established in the CANOPY clinical trial and that the calculated serum neutralizing antibody titers against JN.1 were consistent with the titer levels associated with efficacy in prior clinical trials of adintrevimab (ADG20), the parent mAb for VYD222, and other monoclonal antibody products. JN.1 is currently the dominant variant circulating in the U.S. According to estimates from the Centers for Disease Control and Prevention (CDC).4 PEMGARDA (pemivibart) (4500 mg) is administered as an intravenous (IV) infusion.
PEMGARDA is Invivyd's first authorized mAb and the first mAb to receive EUA based on a rapid immunobridging trial design that is expected to be repeatable to help address the need to mitigate ongoing viral evolution. It was developed using INVYMAB™, the company's platform approach which combines state-of-the-art viral surveillance and predictive modeling with advanced antibody engineering. INVYMAB is designed to enable the rapid, serial generation of durable mAbs targeting conserved epitopes that could be deployed to keep pace with SARS-CoV-2 viral evolution or other viral threats. With a commitment to serial innovation, Invivyd aims to ensure that vulnerable populations, such as immunocompromised people, have continuous access to innovative antibody therapies.
The Company estimates it had approximately $200.6 million of cash and cash equivalents as of December 31, 2023. The estimated amounts are preliminary, have not been audited and are subject to change upon completion of the Company's audited financial statements for the year ended December 31, 2023. In February 2024, the Company sold shares of common stock totaling $40.5 million in gross proceeds under its At-the-Market facility further strengthening the Company's balance sheet ahead of PEMGARDA launch. Based on current operating plans and excluding anticipated cash collections from PEMGARDA sales, Invivyd expects its existing total cash and cash equivalents will enable the company to fund its operating expenses and capital expenditure requirements into the fourth quarter of 2024.
Interim CANOPY Clinical Data Update
CANOPY is an ongoing Phase 3 clinical trial of VYD222 (PEMGARDA) for the pre-exposure prophylaxis of COVID-19 which enrolled adults ≥18 years of age in two cohorts. Cohort A is a single-arm, open-label trial in adults who have moderate-to-severe immune compromise (n=306); Cohort B is a 2:1 randomized, placebo-controlled trial in which adults who do not have moderate-to-severe immune compromise received VYD222 (n=317) or placebo (n=162). The interim data presented below are subject to further analysis.
Summary of CANOPY immunobridging data
An immunobridging approach was used in the CANOPY clinical trial, utilizing the relationship between serum virus neutralizing antibody (sVNA) titers and clinical efficacy that was demonstrated in the previous EVADE clinical trial of adintrevimab (ADG20), the parent mAb for VYD222, and clinical trials of other mAbs that were previously authorized by the FDA. EVADE was a Phase 2/3 randomized, double-blind, placebo-controlled clinical trial of adintrevimab for PrEP and post-exposure prophylaxis of symptomatic COVID-19 in SARS-CoV-2 naïve, unvaccinated individuals, which showed that a neutralizing titer of 3514 on Day 90 was associated with approximately 70% clinical efficacy in the PrEP cohort (approximately 70% relative risk reduction in development of symptomatic COVID-19 between the adintrevimab and placebo arms).
The CANOPY trial was designed to utilize current relevant SARS-CoV-2 variants in the analyses of neutralizing titers. The primary immunobridging endpoint for Cohort A was based on calculated sVNA titers on Day 28 following VYD222 administration compared with the calculated Day 28 reference titer derived from historical Day 90 data from the EVADE trial. The most relevant SARS-CoV-2 variant circulating in the U.S. At the time of the analysis (JN.1), was selected as the variant for the analysis of the primary immunobridging endpoint.
Summary of initial CANOPY immunobridging data from Cohort A (immunocompromised cohort):
Figure 1. Calculated sVNA titers against JN.1 based on observed pharmacokinetic concentration by timepoints (Cohort A)
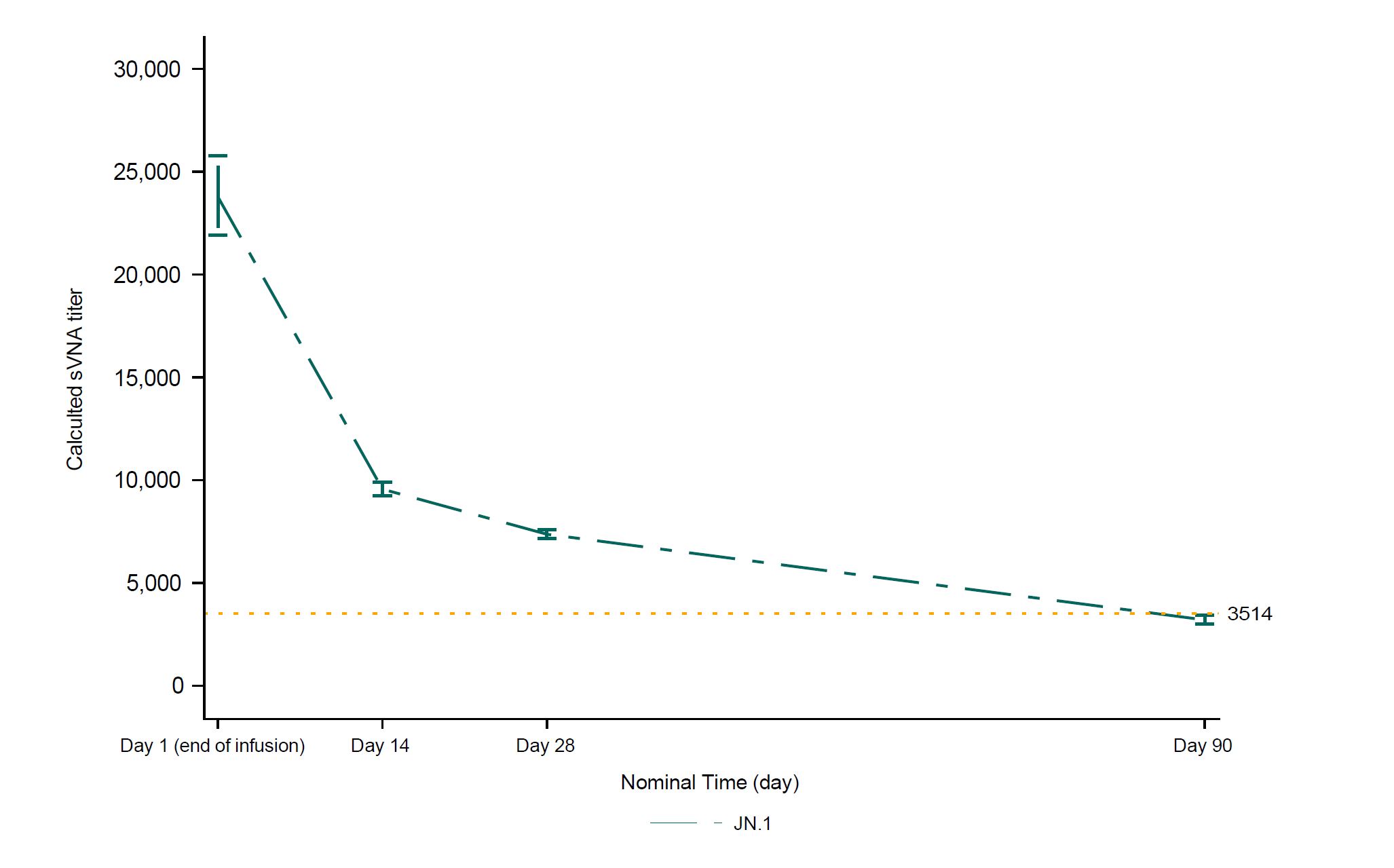 Figure 1 is available at https://www.Globenewswire.Com/NewsRoom/AttachmentNg/bccd1b05-c199-4795-915c-fce5cbe3d651
Figure 1 is available at https://www.Globenewswire.Com/NewsRoom/AttachmentNg/bccd1b05-c199-4795-915c-fce5cbe3d651
Summary of CANOPY safety data
The safety of VYD222 (PEMGARDA) is based on exposure of 623 participants who received at least one dose of VYD222 4500 mg IV in one of two cohorts in the ongoing CANOPY trial. Cohort A is a single-arm, open-label trial in adults who have moderate-to-severe immune compromise including complex underlying medical conditions (n=306). Cohort B is a randomized, placebo-controlled cohort that recruited adults without moderate-to-severe immune compromise who are at risk of acquiring SARS-CoV-2 due to regular unmasked face-to-face interactions in indoor settings. Cohort B participants were randomized 2:1 to VYD222 (n=317) or placebo (n=162). Interim safety data presented today included 296 people in Cohort A who received a second dose of VYD222 three months after the initial dose. In Cohort B, 450 participants received a second dose of VYD222 or placebo three months after the initial dose. Cumulative safety with the first two doses of VYD222 is assessed only in Cohort A because unblinded safety data in Cohort B were not available after Day 28.
Anaphylaxis was observed in four of 623 (0.6%) participants in CANOPY, all in Cohort A. Two participants had anaphylaxis during the first infusion, for whom treatment included diphenhydramine. Two participants had anaphylaxis during the second infusion. All four reactions led to permanent discontinuation of VYD222. Three participants had complete resolution, and one participant had acute resolution with sequelae related to a flare of an underlying condition. For the two participants who experienced anaphylaxis with the second dose, both incidents were reported as life-threatening, and they experienced symptoms during the infusion and following discontinuation of the infusion. Both participants were treated with diphenhydramine and epinephrine. One participant also received oral prednisone and metoprolol for an associated flare of an underlying condition. Please see PEMGARDA Important Safety Information below, including a boxed warning for anaphylaxis.
The systemic infusion-related reactions and hypersensitivity reactions observed in Cohort A are summarized in Table 1. The severity of the reactions was generally mild (17/27) or moderate (8/27), but two reactions were life-threatening.
Table 1. Cohort A (Open-label cohort with moderate-to-severe immune compromise) – Systemic infusion-related reactions and hypersensitivity reactions
Cohort A (n=306) VYD222 First Dose VYD222 First & Second Dose, Cumulatively Systemic infusion-related and hypersensitivity reactions 20 total (20/306 = 7%)(20 mild or moderate,including 2 anaphylaxis*)
27 total (27/306 = 9%)(17 mild and 8 moderate, including 2 anaphylaxis*; plus 2 life-threatening anaphylaxis)
*These two events were initially classified as mild or moderate hypersensitivity adverse reactions. Subsequently, during the review of the EUA application, the FDA reclassified these hypersensitivity adverse reactions as anaphylaxis adverse reactions.
Safety data through Day 28 from Cohort B (post-first dose only) were also analyzed in support of the EUA filing, including randomized data on systemic infusion-related and hypersensitivity reactions, as shown in Table 2. As of Day 28, there were no observations of anaphylaxis in the Cohort B VYD222 arm. Unblinded safety data in Cohort B were not available yet after Day 28.
Table 2. Cohort B (Randomized, placebo-controlled cohort without moderate-to-severe immune compromise at risk of acquiring SARS-CoV-2 due to regular unmasked face-to-face interactions) – Systemic infusion-related reactions and hypersensitivity reactions
Cohort B (n=479) VYD222 First Dose (n=317) Placebo First Dose (n=162) Systemic infusion-related and hypersensitivity reactions 4 total (4/317 =1%)(3 mild and 1 moderate)
0 totalOther than systemic infusion-related reactions and hypersensitivity reactions described previously for Cohort A, the most common (≥2%) treatment-emergent adverse events in Cohort A across both the first and second dose cumulatively, irrespective of causality, observed with VYD222 in participants who have moderate-to-severe immune compromise in CANOPY were upper respiratory tract infection (6%), infusion site infiltration/extravasation/vein rupture (5%), viral infection (4%), influenza-like illness (3%), fatigue (3%), headache (2%), nausea (2%), and local infusion site reactions (2%).
This press release features downloadable multimedia content. View the full suite of assets here: https://www.Multivu.Com/players/English/9254151-invivyd-announces-fda-authorization-pemgarda-formerly-vyd222-pre-exposure-prophylaxis-covid-19/
Conference Call & Webcast
Invivyd will host a conference call and webcast today, Friday, March 22 at 4pm ET. A live audio webcast will be available at https://investors.Invivyd.Com/. Listeners can register for the webcast via this link. Analysts wishing to participate in the question-and-answer session should use this link. A replay of the webcast will be available in the investor section of the company's website approximately two hours after the end of the call. Those who plan on participating are advised to join 15 minutes prior to the start time.
IMPORTANT SAFETY INFORMATION
WARNING: ANAPHYLAXIS
CONTRAINDICATIONS
PEMGARDA is contraindicated in individuals with previous severe hypersensitivity reactions, including anaphylaxis, to any component of PEMGARDA.
WARNINGS AND PRECAUTIONS
Hypersensitivity Including Anaphylaxis and Infusion-Related ReactionsSerious hypersensitivity reactions, including anaphylaxis, have been observed with PEMGARDA. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue administration, and initiate appropriate medications and/or supportive therapy. Clinically monitor individuals during infusion and observe for at least two hours after infusion is complete.
Risk of Cross-Hypersensitivity With COVID-19 VaccinesPEMGARDA contains polysorbate 80, which is in some COVID-19 vaccines and is structurally similar to polyethylene glycol (PEG), an ingredient in other COVID-19 vaccines. For individuals with a history of severe hypersensitivity reaction to a COVID-19 vaccine, consider consultation with an allergist-immunologist prior to PEMGARDA administration.
Risk for COVID-19 Due to SARS-CoV-2 Viral Variants Not Neutralized by PEMGARDACertain SARS-CoV-2 viral variants may emerge that are not neutralized by monoclonal antibodies such as PEMGARDA. PEMGARDA may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants. Inform individuals of the increased risk, compared to other variants, for COVID-19 due to emergent SARS-CoV-2 viral variants not neutralized by PEMGARDA. If signs and symptoms of COVID-19 occur, advise individuals to test for COVID-19 and seek medical attention, including starting treatment for COVID-19 as appropriate.
ADVERSE REACTIONSThe most common adverse events (all grades, incidence ≥2%) observed in participants who have moderate-to-severe immune compromise treated with PEMGARDA included systemic and local infusion-related or hypersensitivity reactions, upper respiratory tract infection, viral infection, influenza-like illness, fatigue, headache, and nausea.
USE IN SPECIFIC POPULATIONS
PregnancyThere are insufficient data to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. PEMGARDA should only be used during pregnancy if the potential benefit outweighs the potential risk for the mother and the fetus.
LactationThere are no available data on the presence of PEMGARDA in human or animal milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PEMGARDA and any potential adverse effects on the breastfed infant from PEMGARDA.
Pediatric UsePEMGARDA is not authorized for use in pediatric patients less than 12 years of age or weighing less than 40 kg. The safety and effectiveness of PEMGARDA has not been established in pediatrics.
EMERGENCY USE AUTHORIZATION (EUA) FOR PEMGARDA
The U.S. Food and Drug Administration (FDA) has issued an EUA for the emergency use of the unapproved product PEMGARDA for the pre-exposure prophylaxis of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg):
LIMITATIONS OF AUTHORIZED USE
PEMGARDA may only be prescribed for an individual patient by physicians, advanced practice registered nurses, and physician assistants that are licensed or authorized under state law to prescribe drugs.
PEMGARDA has been authorized by FDA for the emergency use described above.
PEMGARDA is not FDA-approved for any use, including use for pre-exposure prophylaxis of COVID-19.
PEMGARDA is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of PEMGARDA under Section 564(b)(1) of the Federal Food Drug, and Cosmetic Act, 21 U.S.C. § 360bbb 3(b)(1), unless the authorization is terminated or revoked sooner.
See full Fact Sheet for Healthcare Providers and Fact Sheet for Patients, Parents, and Caregivers for examples of medical conditions or treatments that may result in moderate to severe immune compromise and an inadequate immune response to COVID-19 vaccination, the justification for emergency use of drugs during the COVID-19 pandemic, information on available alternatives, and additional information on COVID-19. The FDA Letter of Authorization is also available for reference.
The prescribing healthcare provider and/or the provider's designee is/are responsible for mandatory reporting of all serious adverse events* and medication errors potentially related to PEMGARDA within 7 calendar days from the healthcare provider's awareness of the event, using FDA Form 3500 (for information on how to access this form, see below). The FDA requires that such reports, using FDA Form 3500, include the following:
Submit serious adverse event and medication error reports using FDA Form 3500 to FDA MedWatch using one of the following methods:
In addition, please provide a copy of all FDA MedWatch forms to:
Invivyd, Inc.Email: pv@invivyd.ComOr call Invivyd, Inc. At 1-800-890-3385 to report serious adverse events.
The prescribing healthcare provider and/or the provider's designee is/are responsible for mandatory responses to requests from FDA for information about serious adverse events and medication errors following receipt of PEMGARDA.
*Serious adverse events are defined as:
You may report side effects related to Invivyd, Inc. Products by sending an email to medinfo@invivyd.Com.
About PEMGARDA
PEMGARDA (pemivibart), formerly known as VYD222, is a half-life extended investigational monoclonal antibody (mAb). PEMGARDA was engineered from adintrevimab, Invivyd's investigational mAb that has a robust safety data package and demonstrated clinically meaningful results in global Phase 2/3 clinical trials for both the prevention and treatment of COVID-19. PEMGARDA was designed for broad activity and has demonstrated in vitro neutralizing activity in pseudotyped virus-like particle and authentic virus neutralization assays against major SARS-CoV-2 variants, including JN.1, the dominant variant in the U.S. Currently according to estimates from the Centers for Disease Control and Prevention. PEMGARDA targets the SARS-CoV-2 spike protein receptor binding domain (RBD), thereby inhibiting virus attachment to the human ACE2 receptor on host cells.
PEMGARDA (pemivibart) injection (4500 mg), for intravenous use is an investigational mAb with emergency use authorization in the U.S. For the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2. Anaphylaxis has been observed with PEMGARDA and the PEMGARDA Fact Sheet for Healthcare Providers includes a boxed warning for anaphylaxis. The most common adverse events (all grades, incidence ≥2%) observed in participants who have moderate-to-severe immune compromise treated with PEMGARDA included systemic and local infusion-related or hypersensitivity reactions, upper respiratory tract infection, viral infection, influenza-like illness, fatigue, headache, and nausea.
About Invivyd
Invivyd, Inc. (Nasdaq: IVVD) is commercial-stage company on a mission to rapidly and perpetually deliver antibody-based therapies that protect vulnerable people from the devastating consequences of circulating viral threats, beginning with SARS-CoV-2. The company's proprietary INVYMAB™ platform approach combines state-of-the-art viral surveillance and predictive modeling with advanced antibody engineering. INVYMAB is designed to facilitate the rapid, serial generation of new monoclonal antibodies (mAbs) to keep pace with evolving viral threats. In March 2024, Invivyd received emergency use authorization (EUA) from the U.S. FDA for its first mAb in a planned series of innovative antibody candidates. Visit https://invivyd.Com/ to learn more.
References
Cautionary Note Regarding Forward Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "anticipates," "believes," "could," "expects," "estimates," "intends," "potential," "projects," and "future" or similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements include statements concerning, among other things, the potential of PEMGARDA as a mAb for pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents who have moderate-to-severe immune compromise; the company's plans related to the commercialization of PEMGARDA, including its expectations regarding availability and supply of PEMGARDA; the ability of the company's INVYMAB platform approach to enable the rapid, serial generation of durable mAbs targeting conserved epitopes that could be deployed to keep pace with SARS-CoV-2 viral evolution or other viral threats; the company's ongoing research and clinical development efforts and future plans, and the timing thereof; the company's expectation that PEMGARDA is the first mAb in a planned series of innovative antibody candidates; the potential repeatability of an immunobridging trial design for mAb candidates to help address the need to mitigate ongoing viral evolution; the company's commitment to ongoing process improvement while working with global regulatory agencies with the aim to increase the speed and efficiency of new mAb candidate development; the future of the COVID-19 landscape, particularly for vulnerable populations; the company's aim to ensure vulnerable populations have continuous access to innovative antibody therapies; the ongoing in vitro neutralizing activity of PEMGARDA against major SARS-CoV-2 variants; the company's mission to rapidly and perpetually deliver antibody-based therapies that protect vulnerable people from the devastating consequences of circulating viral threats, beginning with SARS-CoV-2; the company's preliminary estimate of its cash and cash equivalents balance as of December 31, 2023; the anticipated timeline of the company's cash runway; the company's business strategies and objectives; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company's forward-looking statements and you should not place undue reliance on the company's forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause the company's actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: how long the EUA granted by the FDA for PEMGARDA will remain in effect and whether the EUA is revoked or revised by the FDA; the company's ability to build and maintain sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; changes in expected or existing competition; the timing and progress of the company's discovery, preclinical and clinical development activities; the outcome of the company's engagement with regulators regarding mAb candidate development; whether the company is able to utilize an immunobridging trial design for future mAb candidates; the uncertainties and timing of the regulatory authorization or approval process, and available development and regulatory pathways for authorization or approval of the company's product candidates; changes in the regulatory environment; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of PEMGARDA or any other product candidate following regulatory authorization or approval; the predictability of clinical success of the company's product candidates based on neutralizing activity in preclinical studies; the risk that results of preclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; the company's reliance on third parties with respect to virus assay creation and product candidate testing and with respect to its clinical trials; variability of results in models used to predict activity against SARS-CoV-2 variants; whether PEMGARDA or any other product candidate is able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; the complexities of manufacturing mAb therapies; the company's dependence on third parties to manufacture, label, package, store and distribute clinical and commercial supplies of its product candidates; whether the company is able to provide sufficient commercial supply of PEMGARDA to meet market demand; whether the company can obtain and maintain third-party coverage and adequate reimbursement for PEMGARDA or any other product candidate; the company's ability to leverage its INVYMAB platform approach to enable the rapid, serial generation of durable mAbs that keep pace with SARS-CoV-2 viral evolution or other viral threats; any litigation and other proceedings or government investigations relating to the company; any change in the preliminary estimate of the company's cash and cash equivalents balance as of December 31, 2023 upon completion of the company's audited financial statements for the year ended December 31, 2023; the company's ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company's actual results to differ materially from those expressed or implied in the forward-looking statements in this press release are described under the heading "Risk Factors" in the company's Annual Report on Form 10-K for the year ended December 31, 2022 filed with the Securities and Exchange Commission (SEC), and in the company's other filings with the SEC, and in its future reports to be filed with the SEC and available at www.Sec.Gov. Forward-looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law.
This press release contains hyperlinks to information that is not deemed to be incorporated by reference in this press release.
Contacts:
Media Relations(781) 208-1747media@invivyd.Com
Investor Relations(781) 208-1747investors@invivyd.Com

![]()
Minnesota Republican Calls Vaccines 'poison' In Committee Hearing
ST. PAUL — A Minnesota legislator spoke out against allowing child care facilities to set their own policies regarding immunizations, calling vaccines poison, during a committee hearing Tuesday, March 19.
Rep. Natalie Zeleznikar, R-Fredenberg Township, commented after a proposed bill, HF367, was presented in the House Children and Families Finance and Policy Committee this week. It seeks to give child care providers the option to adopt an immunization policy that would not allow for a conscientiously held belief exemption.
Under Minnesota statute, anyone over 2 months old must be immunized to enroll in schools or child care with an exemption for medical reasons, immunity and conscientious objections.
Zeleznikar objected to the bill and over several minutes told committee members that vaccines are poison, that the bill would negatively affect the workforce in northern Minnesota and she called into question the effectiveness of herd immunity.
"Last year we passed a cannabis bill. There's many people that want to do that because it grows from the ground. It's natural," she said during Tuesday's hearing. "And yet now we're saying to families, by the way, you don't have a choice for a poison that could kill you."
As evidence of her claims, Zeleznikar sent information about vaccines from the Centers for Disease Control and Prevention, which lists possible side effects. On its site, the CDC states there is a very remote chance of a vaccine causing a severe allergic reaction, other serious injury or death.
However, vaccines can prevent infectious diseases that once killed or harmed people, according to the CDC, which says that without vaccines, children are at risk of getting ill or dying from diseases like measles and whooping cough.
The CDC also instructs the public to tell their health care provider about any possible severe, life-threatening allergies.
Zeleznikar acknowledged the low risk of vaccine injuries and said Minnesota has a good track record for childhood vaccination rates when asked to provide context about her statements for this story.
"I worked in healthcare my entire career, and I am not anti-vaccine," Zeleznikar wrote in a statement to Forum News Service. "In fact, I have received vaccinations and had my own kids vaccinated. With that said, as a State Representative, I respect the importance for informed consent for medications, medical procedures, and vaccinations."
Zeleznikar also told committee members Tuesday that since the bill would still allow for medical exemptions, conscientious exemptions should stay in place.
"We're not going to change the risk factor for any of these children at the child care center because we already have some we're going to allow to be there for a medical exemption," she said. "So this is just further going to complicate the issues."
Zeleznikar, whose district covers several communities north of Duluth, said parents should have the right to choose whether their children should be vaccinated and that not allowing for conscientious objection could open up legal questions for excluding religious exemptions.
"Conscientious exemptions encompass a family's religious beliefs or conscientious beliefs and are utilized when their beliefs prevent them from accepting one or more of the vaccines doses," she wrote in her statement. "It allows space for public health recommendations while respecting the diverse population and individual autonomy of all citizens."
Current Minnesota statute and the proposed bill do not prevent parents from making that decision.
"This bill would simply allow each child care provider to make their own decision about accepting exemptions to vaccines only in the case of parental preference," bill author Rep. Mike Freiberg, DFL-Golden Valley, said. "Child care providers are private businesses and should be allowed to choose the clients they serve, within the bounds of anti-discrimination laws of course, and vaccine status is not a protected class."
Most vaccine side effects are mild and include soreness at the injection site, fussiness or a low-grade fever, according to the CDC.
Vaccines are so effective against harmful and deadly diseases partly due to herd immunity, which calls for the vast majority of a population to be immunized to protect people who cannot be vaccinated, like newborns or those with compromised immune systems, according to the CDC.
Zeleznikar's comments have drawn scrutiny from the Democratic-Farmer-Labor Party.
"Natalie Zeleznikar has repeatedly proven that she cares more about pushing vaccine conspiracy theories and fighting culture wars than she does about delivering for her district," DFL Party Chair Ken Martin said. "Her constituents deserve a legislator that will prioritize governing and improving their lives, and they will hold her accountable for embracing conspiracy theories while taking far-right votes against everything from paid family leave to free school meals."
Rep. Kim Hicks, DFL-Rochester, spoke during Tuesday's hearing against Zeleznikar's comments. Hicks' daughter has had adverse effects against vaccines and has not been able to follow a normal vaccine schedule.
"As a parent of a child who has real, very dangerous, very, very scary medical conditions, I am insulted that that is somehow equivalated similarly. It's not. It's not the same," Hicks said. "This bill protects my daughter who could not be vaccinated."
Before the committee hearing, Zeleznikar appeared alongside Rep. Krista Knudsen, R-Lake Shore, in a Facebook video where the pair spoke about their support for opposition to childhood vaccines.
Zeleznikar said in the video that it seems logical that a parent could opt out of vaccinating their child.
In addition to Zeleznikar and Knudsen's criticism of vaccines, Rep. Walter Hudson, R-Albertville, spoke against the proposed bill to give child care facilities a broader choice regarding their immunization policies.
He denounced earlier testimony during the hearing that said the bill would lead to higher vaccination rates, calling it forced coercion, even though the bill neither requires parents to vaccinate their children nor forces child care centers to adopt a policy that would bar conscientious objections from vaccine requirements.
"I can tell I'm not winning anybody over with this and that's fine," he said during the hearing. "But I hope that at the very least there is some degree of respect for the fact that people are entitled to control their own bodies."
Comments
Post a Comment